What is Gaucher Disease (Glucocerebroside Lipidosis, Glucocerebrosidosis)?
Gaucher disease (glucocerebrosid lipidosis, glucocerebrosidosis) is one of the most frequent inherited disorders of glycolipid metabolism.
It is believed that Gaucher disease is based on allelic mutations of the genes encoding glucocerebrosidase, an enzyme that catalyzes the hydrolytic detachment of glucose from glucocerebroside, which accumulates in various organs and tissues. For the first time a patient with this pathology was described by the French dermatologist Gaucher (Ph. Ch. E. Gaucher) in 1882.
Causes of Gaucher Disease (Glucocerebroside Lipidosis, Glucocerebrosidosis)
The type of inheritance is autosomal recessive.
Pathogenesis during Gaucher Disease (Glucocerebroside Lipidosis, Glucocerebrosidosis)
A deficiency of the enzyme beta-glucocerebrosidase, catalyzing the removal of glucose from glucocerebroside, was found. This leads to the accumulation of cerebrosides and their deposition in the cells of the reticuloendothelial system.
Symptoms of Gaucher Disease (Glucocerebroside Lipidosis, Glucocerebrosidosis)
There are three clinical types that differ depending on the age at which the disease began, and the prevalence of one or another symptom. It occurs in all ethnic groups, but there is an increased incidence of type I disease among Ashkenazi Jews (1: 25,000 newborns). Types II and III are observed mainly in non-Jewish populations.
In all clinical forms, there is a decrease in the activity of the enzyme glucocerebrosidase in various organs and tissues. There is a dependence of the severity of the clinical picture on the degree of decrease in the activity of glucocerebrosidase. In type II, the enzyme activity does not exceed 5%; in type I, it varies from 18 to 40%. The source of accumulating glucocerebroside is leukocytes and red blood cells. Excessive enlargement of the liver and spleen is explained in addition to the accumulation of glucocerebroside by the compensatory reaction of the cells in order to obtain an additional amount of glucocerebrosidase. Cells filled with glucocerebroside with an eccentrically displaced nucleus and cytoplasm resembling crumpled tissue paper or meringues are called Gosha cells. They are found in the lymph nodes, alveoli capillaries, bone marrow, in the adventitia of arterioles, veins, lymphatic vessels. In the histopathological picture of the central nervous system, the death of neurons prevails, Gaucher cells in the affected area are rare.
Type I (chronic, or adult, neuropathic) is characterized by the development of splenomegaly shortly after birth. In some cases, the phenomenon of hypersplenism with thrombocytopenia, anemia, leukopenia, increased bleeding. Hepatomegaly is detected a little later. Bone disorders include seizures of bone pain that last for days and weeks, fractures, expansion of part of the femur, and aseptic necrosis of its head. These symptoms are based on a strong expansion of the bone marrow cavity and an increase in the mass of cells accumulating glucocerebroside. Infiltration by the macrophages of the lungs and heart is possible, leading to pulmonary hypertension and susceptibility to the development of pneumonia. In some patients at a later age, yellow-brown pigmentation of the face and lower extremities is noted. Often find yellow-brown areas of a thickening of a cornea. Neurological symptoms are absent. Patients live to middle age, although life expectancy varies greatly. The course of the disease is slow and progressive.
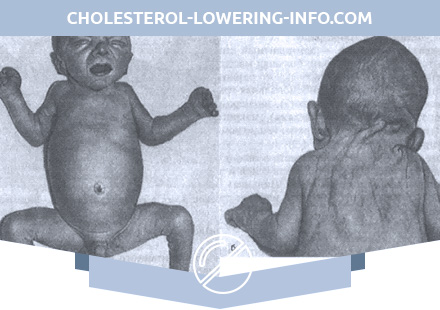
Type II (acute infantile, acute neuropathic) can develop immediately after birth and up to 18 months (mostly in 3 months). Initial manifestations – weak cry, sluggish sucking, lack of Moro reflex, hepatosplenomegaly. In children, strabismus is detected, swallowing is difficult, the head is in the retroflection position. By 9 months, spastic paralysis develops with elevated tendon reflexes, clonus, and muscle stiffness. In rare cases, there are convulsions. More and more progressive bulbar disorders lead to the death of a child as a result of repeated pneumonia at the age from 1 month to 2 years (average age is 9 months).
Type III (juvenile, or subacute, neuropathic) is manifested in the second year of life by splenomegaly. Hepatomegaly is moderately expressed. This is followed by a “period of silence” lasting from 3 to 7 years, followed by obvious neurological disorders. Neurological symptoms (strabismus, spastic paralysis, ataxia, convulsions) are not pronounced. Death usually occurs 2 years after the appearance of neurological disorders.
Diagnosis of Gaucher Disease (Glucocerebroside Lipidosis, Glucocerebrosidosis)
The diagnosis is made on the basis of the clinical picture, the detection of Gaucher cells in the bone marrow, liver and lymph nodes, the quantitative determination of glucocerebroside in tissue biopsies and the direct analysis of glucocerebrosidase activity in leukocytes, liver cells and spleen. The diagnosis is confirmed by x-ray examination of the bones, which allows to identify the expansion of the cortical layer of the distal femur (Erlenmeyer cones).
The differential diagnosis is carried out with Niemann-Pick’s disease, in which the enlargement of the spleen is detected later than hepatomegaly, there is a pathology of vision and hearing; with GM1-type I gangliosidosis, characterized, in addition to the symptoms, by pronounced phenotype features. The final diagnosis is based on biochemical data.
Treatment of Gaucher Disease (Glucocerebroside Lipidosis, Glucocerebrosidosis)
Patients should be monitored by a hematologist throughout their lives. In some cases, splenectomy is indicated. Bone pain requires the use of analgesics, orthopedic advice. In type I, intravenous administration of purified glucocerebrosidase leads to a decrease in its deposits in the liver; The possibility of reversibility of clinical manifestations with the introduction of the enzyme in liposomes is being studied. Methods of treatment of the other two types are not proposed.
Prevention of Gaucher Disease (Glucocerebroside Lipidosis, Glucocerebrosidosis)
Medical genetic counseling and examination in a specialized clinic to identify heterozygous carriers (determine the activity of glucocerebrosidase in leukocytes and fibroblasts using labeled glucocerebroside). Prenatal diagnosis is possible by examining glucocerebrosidase activity in cultured amniotic cells.